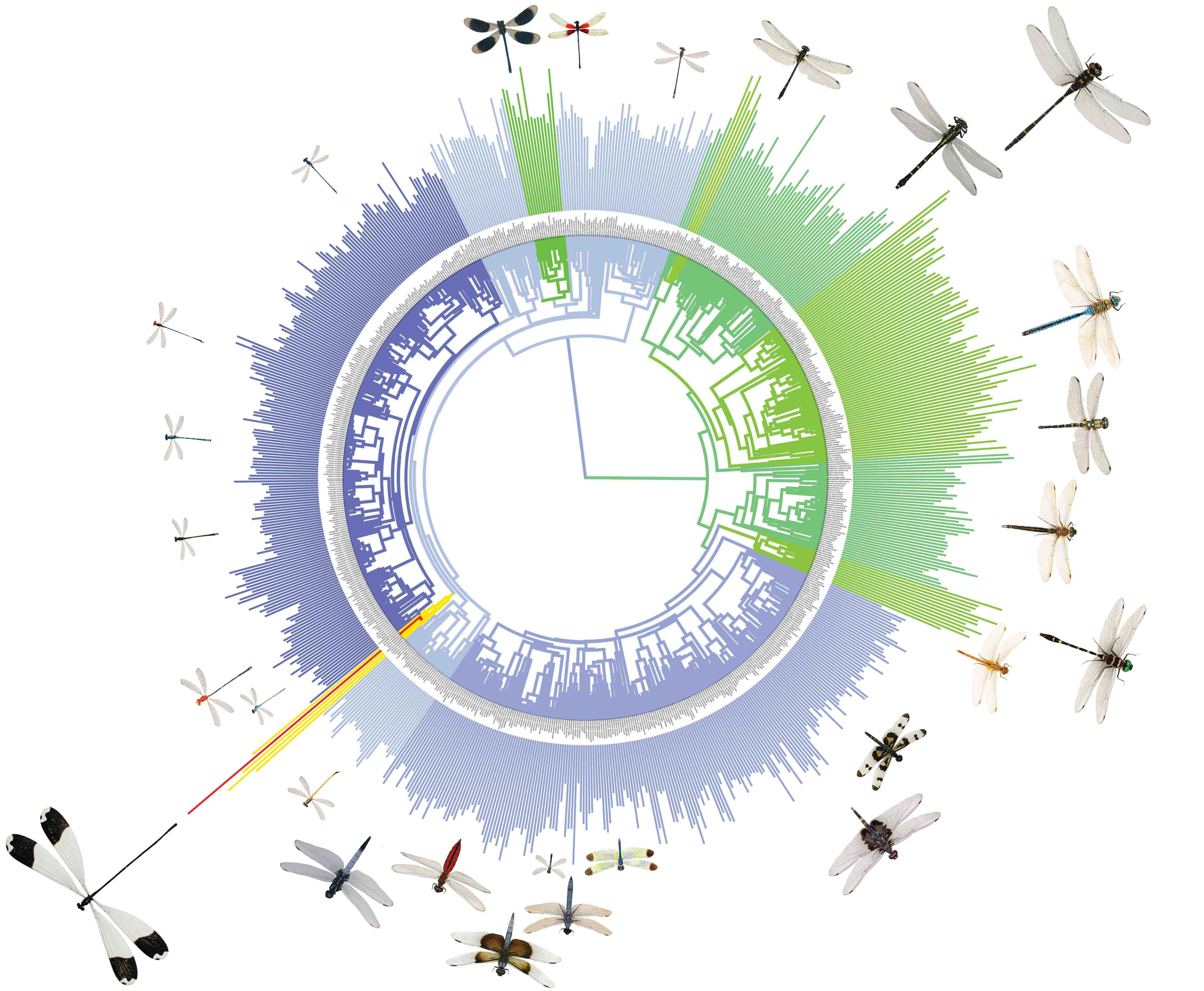
The Odonate "Super" Tree is a large tree of damselfies and dragonflies built from sequences on GenBank.
The tree is comprised 1322 taxa, which is around 21% of known odonates (n = 6400). A total of 759 of these taxa were from the suborder Zygoptera (damselflies) and a total of 563) from Anisoptera (dragonflies). All 32 known odonate families are represented in this phylogeny.
The Tree is meant to be used with the Odonate Phenotypic DataBase.
More information can be found below and in the original article "Body size evolution in an old insect order".
The tree is time calibrated with fossils dowloaded from the Paleobiology Database. We considered taxonomic groups and names found on the World Odonata List. We queried GenBank for each species in the list and then downloaded all of the associated data using the R package seqinr. Annotations were then processed and cleaned of extraneous text to determine the downloaded genetic region. Our phylogeny was constructed using BEAST v1.8.1.Two separate time calibrated trees were generated in BEAST for computational reasons (one for Zygoptera and one for Anisoptera) and they were later combined (using the median age of the root of each subtree) into a single tree with divergence time set between the two suborders with fossils.
The following genetic regions were used in the construction of our trees: tRNA-Val, histone H3, 12S ribosomal RNA, NADH dehydogenase subunit I, tRNA-Leu, cytochrome oxidase II, elongation factor 1 alpha, internal transcribed spacer 2, 5.8S ribosomal RNA, internal transcribed spacer 1, 18S ribosomal RNA, 28S ribosomal RNA, 16S ribosomal RNA, and cytochrome oxidase I.
Cleaned genetic regions were then aligned using the R package muscle and in parallel using the R package foreach. The alignments were processed using gblocks, which eliminates poorly aligned positions. This was done using the gblocks function in the R package phyloch. PartitionFinder was run on each the concatenated alignments to determine the best partitioning scheme for our aligned sequences.
All of the steps above were glued together and automated in R, so it is possible to update the tree as new sequences become available.